
World Sickle Cell Day: Why Sickle Cell Requires Global Attention Even Amidst a Pandemic?
World Sickle Cell Day is a United Nations recognised day to raise awareness of sickle cell at a national and international level. On 22nd December 2008, the United Nations General Assembly adopted a resolution that recognises sickle cell disease as a public health problem and “one of the world’s foremost genetic diseases.” The resolution calls for members to raise awareness of sickle cell on June 19th of each year at the national and international levels.
Despite Sickle Cell Disease (SCD) being recognised by the World Health Organization (WHO) as a global health problem, there is not a coordinated approach to tackling the disease at a global level. As SCD continues to be an under-recognised public health issue, urgent action, attention, and awareness among the global community are required to tackle the disease.
Earlier it was thought that pathophysiology of the clinical complications of SCD was due to the direct entrapment of deformed sickle-shaped RBCs in the microcirculation. But the hypercoagulable state of SCD is due to a multitude of factors like enhanced platelet function, activation of the coagulation cascade, and impaired fibrinolysis, which may also be affected at a greater level due to the sickled RBC structure. All these lead to the formation of thrombus, which could lead to deprivation of oxygen to tissues and organs leading to organ damage. These blockages also can lead to painful crises called Vaso-occlusive Crisis (VOCs), which are considered the clinical hallmark of the disease and the main reason why patients seek medical care in hospitals.
Children and adults with sickle cell disease often require care in the emergency department of hospitals and clinics for health issues related to SCD. The ED may be a patient’s only option for health care when symptoms, such as pain crises, cannot be managed at home or when a patient does not have access to a healthcare provider who specializes in treating SCD. The Sickle Cell Data Collection (SCDC) program found that in California, people with SCD seek care in the ED an average of three times a year from their late teens to their late fifties.
SCD & Tribal Population In India
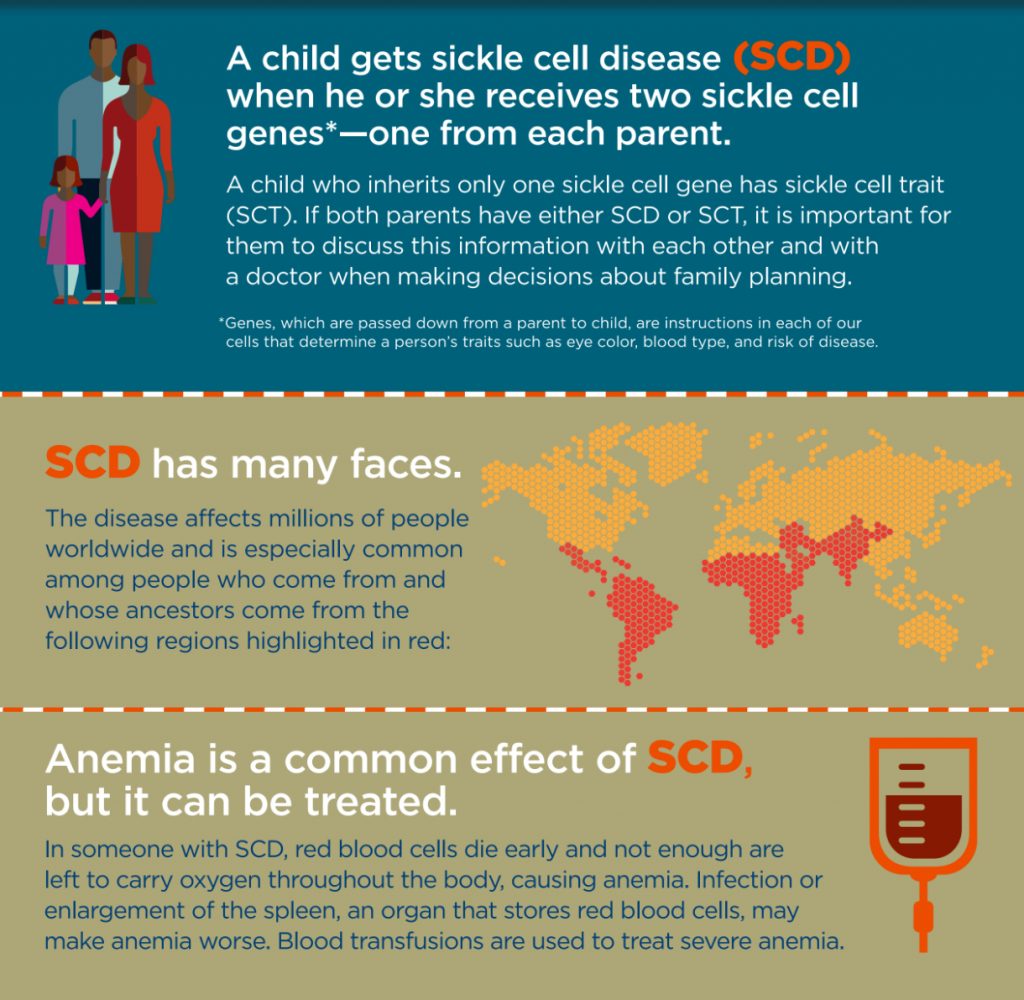
The sickle cell disease is widespread among many tribal population groups in India with the prevalence of heterozygotes varying from 1-40 percent. As per the study published in the Indian Journal of Medical Research (IJMR), Madya Pradesh has the highest load with an estimated number of 961,492 sickle heterozygotes and 67,861 sickle homozygotes. Further, 27 out of the 45 districts in Madhya Pradesh fall under the sickle cell belt and the prevalence of HbS varies from 10 to 33 percent. It has also been estimated that 13,432 pregnancies would be at risk of having a child with sickle cell disease in the state and the expected annual births of sickle homozygotes would be 3358.
In Maharashtra, the sickle gene is widespread in all the eastern districts, also known as the Vidarbha region. The prevalence of sickle cell carriers in different tribes varies from 0 to 35 percent. Similarly, a very high prevalence of HbS (18.2 to 34.1%) is seen among the tribes in the Wayanad district of Kerala. However, the entire tribal population of 125,000 individuals in the area was screened followed by genetic counseling where carriers of HbS were advised not to marry carriers.
Most of the tribal populations, where sickle cell disease is common, rely on the primary health care facilities in rural and often remote areas. Thus, the goals of medical genetic services should be to help these people with a genetic disadvantage to provide access to quality care as well as social and genetic counselling support to make informed choices for reproduction to have healthy children with the availability of prevention programmes when needed. The Indian Council of Medical Research (ICMR) under its Tribal Health Research Forum (THRF) activities as well as other programmes under the National Rural Health Mission (NRHM) in different states have initiated programmes to enable advances in genetics to reach these communities.
Symptoms & Treatment
People with sickle cell disease start to have signs of the disease during the first year of life, usually around 5 months of age. Symptoms and complications of SCD are different for each person and can range from mild to severe.
The reason that infants don’t show symptoms at birth is that the baby or fetal hemoglobin protects the red blood cells from sickling. When the infant is around 4 to 5 months of age, the baby or fetal hemoglobin is replaced by sickle hemoglobin and the cells begin to sickle. SCD is a disease that worsens over time. Treatments are available that can prevent complications and lengthen the lives of those who have this condition. These treatment options can be different for each person depending on the symptoms and severity.

Hydroxyurea is a medicine that can decrease several complications of SCD. This treatment is very safe when given by medical specialists experienced in caring for patients with SCD. However, the side effects of taking hydroxyurea during pregnancy or for a long time are not completely known. The Food and Drug Administration has also approved a new medicine to reduce the number of sickle cell crises in adults and children older than age five; it is called Endari (L-glutamine oral powder).
Another treatment, which can cure SCD, is a stem cell transplant (also called a bone marrow transplant); this procedure infuses stem cells into the body to replace damaged or diseased bone marrow. Although transplants of bone marrow or blood from healthy donors are increasingly being used to successfully cure SCD, they require a matched donor (a person with similar, compatible bone marrow), and transplants can sometimes cause severe side effects, including occasional life-threatening illness or death.
The US Department of Health and Human Services has issued detailed guidelines for evidence-based management of Sickle Cell Disease. The practice guidelines best supported by scientific evidence are:
- Penicillin prophylaxis prevents pneumococcal sepsis in children.
- In surgical settings, simple transfusions to increase hemoglobin (Hb) levels to 10 g/dL are as good as or safer than aggressive transfusions to reduce sickle hemoglobin (Hb S) levels to below 30 percent.
- Pneumococcal vaccine prevents pneumococcal infection in children.
- Transfusions to maintain a hematocrit of more than 36 percent do not reduce complications of pregnancy.
- Transfusions to reduce Hb S levels to below 30 percent prevent strokes in children with high central nervous system blood flow.
- Hydroxyurea decreases crises in patients with severe sickle cell disease.
Sickle Cell & COVID-19
Individuals of any age with blood disorders such as sickle cell disease may be at an increased risk of developing serious COVID-19 symptoms. These individuals should follow general guidelines to keep their immune systems strong.
The U.S. Centers for Disease Control and Prevention (CDC) advises patients living with a chronic disease to stock up on essential medicines and supplies that can last for a few weeks. It also recommends social distancing and avoiding crowds and non-essential travel to prevent the spread of the virus.
There is a high possibility for the Sickle Cell Day to get overshadowed by the COVID-19 pandemic. But the facts and figures certainly prove that SCD as a disease requires similar attention and recognition within the global context.